


Materiał(y) wyjściowy(e) do produkcji substancji czynnej (API) – ważność wyboru
- 4 grudnia 2014
- API, banki komórek, GMP, ICH Q11, Macierzysty Bank Komórek, materiał wyjściowy, MCB, redefiniowanie materiału wyjściowego, Roboczy Bank Komórek, substancja czynna, substrat komórkowy, WCB,
INFORMACJE O WYDARZENIACH ON-LINE


Zachęcamy i serdecznie zapraszamy Wszystkich Państwa oraz Firmy
do uczestnictwa w wydarzeniach ON-LINE
Lhasa Limited jest organizacją not-for-profit dostarczającą firmom rozwiązania w zakresie oprogramowania, które wspomagają podejmowanie świadomych decyzji na temat bezpieczeństwa chemicznego, zmniejszając tym samym potrzebę przeprowadzania testów na zwierzętach, usprawniając długotrwały i kosztowny proces rozwoju nowych leków i zabezpieczając zdrowie ludzkie przed szkodliwym działaniem substancji chemicznych. Zastosowanie Derek Nexus i Sarah Nexus może być pomocne w zakresie spełnienia zgodności z wytycznymi ICH M7 a Mirabilis jest narzędziem analizy ryzyka, które pozwala użytkownikom obliczyć czy potencjalnie mutagenne zanieczyszczenie zostanie wypłukane na drodze syntezy. To z kolei zapewnia przemysłowi standardowe podejście, które jest uznawane przez regulatorów zgodnie z wytycznymi ICH M7. Więcej informacji można znaleźć na stronie internetowej: https://www.lhasalimited.org/.
Zgodnie z dokumentem Część II: Podstawowe wymagania dla substancji czynnych używanych jako materiały wyjściowe do wytycznych Dobrej Praktyki Wytwarzania (GMP) wytwórca określa i dokumentuje racjonalne uzasadnienie wyboru etapu procesu, w którym rozpoczyna się produkcja substancji czynnej.

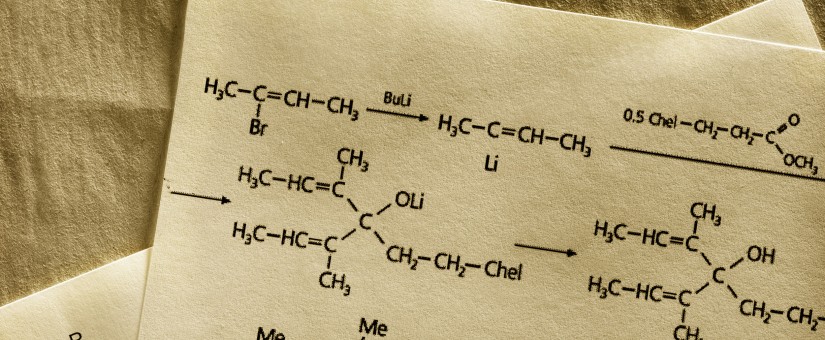
Dokument ten definiuje „Materiał wyjściowy do produkcji substancji czynnej (API)” jako „surowiec, produkt pośredni lub substancję czynną, który(a) jest wykorzystywany(a) do produkcji danej substancji czynnej, i który(a) jest wbudowywany(a) jako znaczący/istotny fragment struktury danej substancji czynnej. „Materiał wyjściowy do produkcji substancji czynnej” może być artykułem komercyjnym (materiałem powszechnie dostępnym), materiałem zakupionym od jednego lub więcej dostawców na podstawie kontraktu lub umowy handlowej, lub też może być wytwarzany wewnętrznie (u wytwórcy danej substancji czynnej). Materiały wyjściowe do produkcji substancji czynnej mają zwykle zdefiniowane własności chemiczne i strukturę”.
Dla procesów syntezy, produkcja substancji czynnej rozpoczyna się w tym etapie procesu, w którym „Materiał wyjściowy do produkcji substancji czynnej” zostaje wprowadzony do procesu. Dla innych procesów (np. fermentacji, ekstrakcji, oczyszczania, itd.), wskazanie etapu procesu, w którym rozpoczyna się produkcja należy określić dla każdego przypadku z osobna.

Dlaczego określenie etapu procesu, w którym rozpoczyna się wytwarzanie substancji czynnej jest tak istotne?

Począwszy od tego etapu, kolejne etapy wytwarzania produktów pośrednich i/lub substancji czynnych należy prowadzić zgodnie z zasadami Dobrej Praktyki Wytwarzania (GMP), które zostały zdefiniowane w Części II: Podstawowe wymagania dla substancji czynnych używanych jako materiały wyjściowe.
Zgodnie ze zmienionym Rozdziałem 5: Produkcja do wytycznych Dobrej Praktyki Wytwarzania (GMP) należy ustanowić system identyfikacji łańcucha dostaw od „Materiałów wyjściowych do produkcji substancji czynnej” po końcowy produkt leczniczy a towarzyszące ryzyka należy formalnie oceniać i okresowo weryfikować.
Wybór materiałów wyjściowych i materiałów źródłowych (termin używany w niektórych regionach w odniesieniu do materiałów wyjściowych dla biotechnologicznych (wytworzonych w procesach biotechnologicznych)/biologicznych substancji czynnych) został szeroko opisany w wytycznej ICH Q11 Guideline on development and manufacture of drug substances (chemical entities and biotechnological/biological entities).
Poniżej podano kilka ogólnych zasad, które należy wziąć pod rozwagę przy wyborze materiałów wyjściowych i materiałów źródłowych (zgodnie z wytyczną ICH Q11):
- dla syntetyzowanych substancji leczniczych
- właściwe kontrole substancji leczniczej i procesu jej wytwarzania (włączając odpowiednie kontrole dla zanieczyszczeń)
- materiał wyjściowy powinien być substancją o zdefiniowanych właściwościach chemicznych i strukturze
- materiał wyjściowy został wbudowany w strukturę substancji leczniczej jako znaczący/istotny fragment strukturalny
- dla półsyntetycznych substancji leczniczych (gdzie składniki struktury zostały wprowadzone poprzez połączenie syntezy chemicznej i elementów pochodzenia biologicznego) (np. uzyskanych z fermentacji lub poprzez ekstrakcję z materiału botanicznego)
- proces wytwarzania może być opisany rozpoczynając od materiału źródłowego (mikroorganizmu lub materiału botanicznego)
- wyizolowany produkt pośredni może (pod pewnymi warunkami) być również zaproponowany jako materiał wyjściowy
- dla biotechnologicznych (wytworzonych w procesach biotechnologicznych)/biologicznych substancji leczniczych (zgodnie z wytycznymi ICH Q5A, Q5B i Q5D)
- banki komórek stanowią początkowy etap w procesie biotechnologicznego wytwarzania substancji leczniczych oraz niektórych biologicznych substancji leczniczych (banki komórek powinny być dobrze zdefiniowane, scharakteryzowane i skwalifikowane; idea dwupoziomowego banku komórek, w której Macierzysty Bank Komórek jest wykorzystywany do utworzenia Roboczych Banków Komórek, jest ogólnie akceptowana jako najlepsze praktyczne podejście odnośnie dostarczania substratu komórkowego do ciągłego wytwarzania produktu; w odniesieniu do bezpieczeństwa wirusologicznego – wymagana jest pełna charakterystyka materiału wyjściowego stanowiącego substrat komórkowy w kierunku określenia obecności i identyfikacji zanieczyszczenia wirusami; w tym miejscu należy podkreślić, że charakterystyka i badania substratów komórkowych pochodzących z banków komórek są krytycznym elementem kontroli produktów biotechnologicznych i biologicznych).



Mogłoby się wydawać, że szeroko opisane rozważania dotyczące wyboru materiałów wyjściowych/źródłowych pomogą w tej kwestii podmiotom odpowiedzialnym, jednakże rzeczywistość okazała się zgoła inna. Zauważono, że z powodu nieprawidłowego zrozumienia i wolnej interpretacji zapisów powyżej wspomnianej sekcji wytycznej Q11, w ostatnim czasie wzrosła liczba rozbieżności pomiędzy podmiotami odpowiedzialnymi i osobami oceniającymi jakość z ramienia agencji, asesorami, odnośnie odpowiedniości proponowanych materiałów wyjściowych.
Aby objaśnić niektóre oczekiwania europejskich kompetentnych organów/władz, które wynikają z wytycznej ICH Q11 a dotyczą informacji, które należy przedłożyć w dossier rejestracyjnym, aby uzasadnić wybór materiałów wyjściowych, EMA opublikowała dokument zatytułowany Reflection paper on the requirements for selection and justification of starting materials for the manufacture of chemical active substances. Dotyczy on substancji leczniczych wytworzonych na drodze syntezy oraz półsyntetycznych substancji leczniczych. Dokument ten powinien zharmonizować opinie osób oceniających oraz doprecyzować wymagania, które ciążą na podmiotach odpowiedzialnych.
Poniżej pokazano kilka przykładów nieścisłości/braków, które zostały zidentyfikowane w złożonej dokumentacji przez osoby ją oceniające:
- proponowanie bardzo krótkich dróg syntezy ze złożonymi materiałami wyjściowymi zsyntetyzowanymi na zamówienie
- wielokrotne i złożone transformacje procesu syntezy przeprowadzane w jednym naczyniu bez izolacji produktów pośrednich („reakcje jednego kotła”)
- termin „znaczący/istotny fragment strukturalny” jest często nieprawidłowo interpretowany przez podmioty i rozumiany jako bardzo zbliżony strukturalnie do substancji czynnej (podobna wielkość i złożoność struktury)
- deklaracje GMP oraz zgłaszanie chęci i gotowości do inspekcji składane przez podmioty/wytwórców, odnośnie wytwarzania materiałów wyjściowych nie są uznawane przez agencje z tego tytułu, że obecnie wytwarzanie „Materiału wyjściowego do produkcji substancji czynnej” jest wyłączone z zastosowania wytycznych Dobrej Praktyki Wytwarzania (GMP)
- stwierdzenie, że materiał jest handlowo dostępny, bez dodatkowych informacji uzasadniających wybór tego materiału, nie może być uznane za wystarczające
- informacje przedłożone przez podmioty odpowiedzialne lub właścicieli/posiadaczy ASMF (Active Substance Master File), uzasadniające wybór materiałów wyjściowych i ich proponowane specyfikacje, są często niewystarczające do właściwej oceny ich odpowiedniości.
Ważne, żeby w tym miejscu dodać, że podobne problemy związane z materiałami wyjściowymi zostały zidentyfikowane przez EDQM po dokonaniu pierwszych ocen nowych aplikacji złożonych w celu uzyskania certyfikatu zgodności z Farmakopeą Europejską (Certificates of Suitability (CEP)) i zostały przedstawione w dokumencie zatytułowanym Top ten deficiencies New Applications for Certificates of Suitability.

Poniżej przedstawiono ranking nieścisłości/braków dotyczących materiałów wyjściowych, które zostały zidentyfikowane przez EDQM:
- brak rozważań/dyskusji na temat przenoszenia zanieczyszczeń/produktów ubocznych, pochodzących z kluczowych materiałów procesowych (materiałów wyjściowych, produktów pośrednich)
- proponowany materiał wyjściowy nie został zaakceptowany
- brak porównania jakości finalnej substancji wytworzonej z materiałów wyjściowych pochodzących od różnych dostawców
- niekompletne specyfikacje zadeklarowanych materiałów wyjściowych.
Przykłady możliwych implikacji spowodowanych niewłaściwym wyborem i uzasadnieniem materiału wyjściowego:
- redefiniowanie materiału wyjściowego (w kierunku wcześniejszego etapu syntezy)
- dostarczenie zaktualizowanych sekcji CTD (Common Technical Document)
- opóźnienia w procedurze rejestracyjnej.